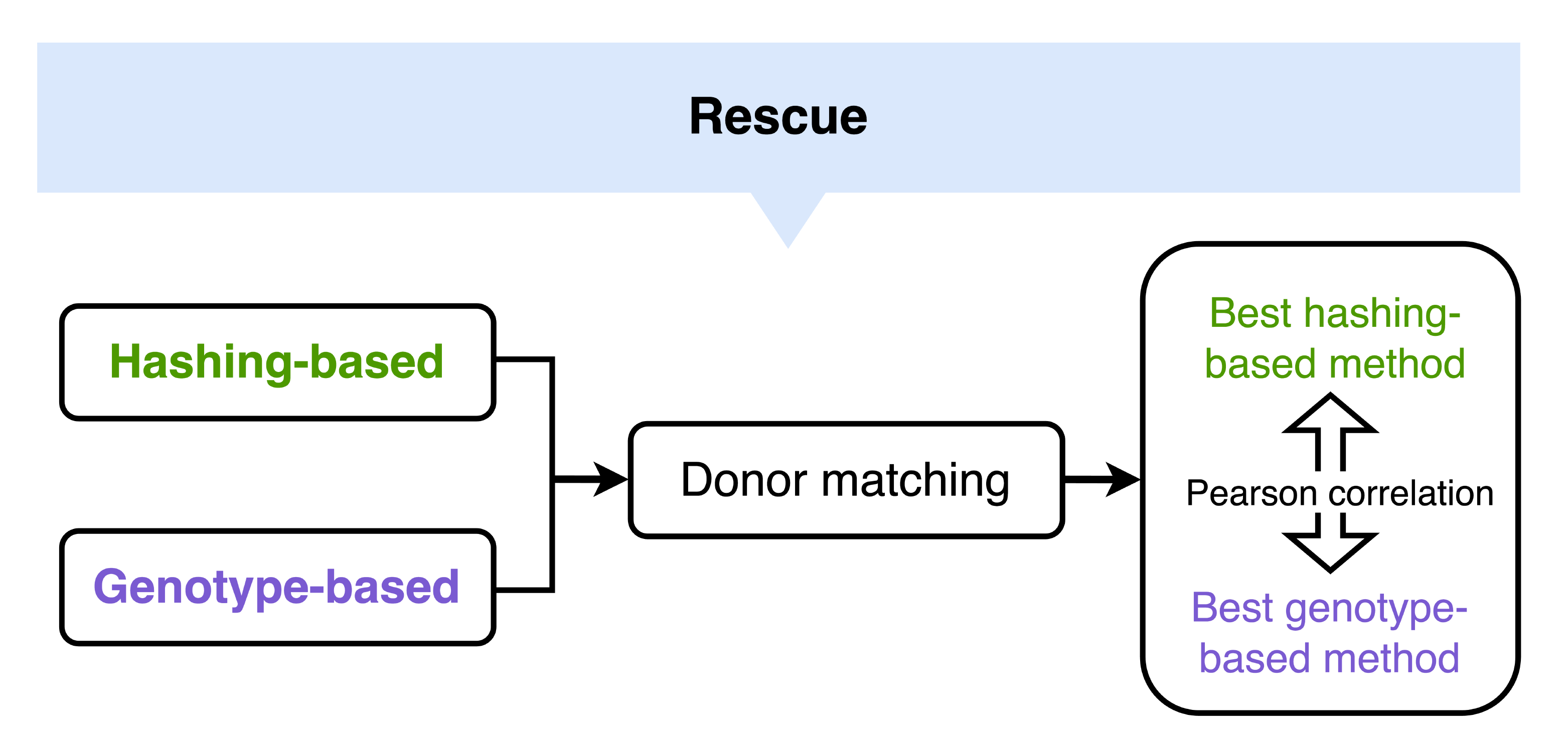
Combining results: rescue mode¶
The joint call of hashing and genetic deconvolution methods has been shown to be beneficial for cell recovery rate and calling accuracy. hadge provides a rescue mode to run both genotype- and hashing-based approaches jointly to rescue problematic hashing experiments in cases where donors are genetically distinct. In this scenario, samples of both hashing and genetic multiplexing experiments are deconvoluted simultaneously. Furthermore, hadge allows for the automatic determination of the best combination of hashing and SNP- based donor deconvolution tools.
Overview¶
Quick start¶
cd hadge
nextflow run main.nf -profile test
Parameter¶
match_donor |
Whether to match donors. Default: True |
demultiplexing_result |
A CSV file with demultiplexing assignment only when running in donor_match mode. In other modes, the input is passed by the pipeline automatically. Default: None |
match_donor_method1 |
The method name to match donors. If None, all genotype-based methods are compared. Default: None |
match_donor_method2 |
The method name to match donors. If None, all hashing-based methods are compared. Default: None |
findVariants |
Whether to extract a subset of informative variants when best genotype-based method for donor matching is vireo. |
variant_count |
The threshold for the minimal read depth of a variant in the cell group when subseting the informative variants by default. Default: 10 |
variant_pct |
The threshold for the minimal frequency of the alternative or reference allele to determine the dominant allele of a variant in the cell group when subseting the informative variants by default. Default: 0.9 |
vireo_parent_dir |
A parent folder which contains the output folder of vireo in the format of |
Output¶
By default, the pipeline is run on a single sample. In this case, all pipeline output will be saved in the folder $projectDir/$params.outdir/rescue. When running the pipeline on multiple samples, the pipeline output will be found in the folder "$projectDir/$params.outdir/$sampleId/rescue.
To simplify this, we’ll refer to this folder as $pipeline_output_folder from now on.
In rescue mode, the genotype- and hashing-based demultiplexing workflow run in parallel. They save their output in $pipeline_output_folder/[gene/hash]_demulti. Before running the donor-matching preocess, the pipeline merges the results of two workflows into classification_all_genetic_and_hash.csv and assignment_all_genetic_and_hash.csv in the $pipeline_output_folder/summary folder.
The following additional output can be found in $pipeline_output_folder/donor_match.
Optional output: Donor matching¶
Folder
[method1]_[task_ID/sampleId]_vs_[method2]_[task_ID/sampleId]with:correlation_res.csv: correlation scores of donor matchingconcordance_heatmap.png: a heatmap visualising the the correlation scoresdonor_match.csv: a map between hashtag and donor identity.all_assignment_after_match.csv: assignment of all cell barcodes after donor matchingintersect_assignment_after_match.csv: assignment of joint singlets after donor matching
General output in the
$pipeline_output_folder/donor_matchfolder:all_assignment_after_match.csv: assignment of all cell barcodes based on the donor matching of the optimal matchdonor_match.csv: a map between hashtags and donor identities based on the donor matching of the optimal matchscore_record.csv: a CSV file storing the matching score and the number of matched donors for each method pair
Optional output: scverse data structures¶
Folder data_output with:
an Anndata object which contains the filtered scRNA-seq counts from
params.rna_matrix_fileredand the assignment of the best-matched method pair after donor matchingan Mudata object which contains the filtered scRNA-seq counts from
params.rna_matrix_fileredand the filtered HTO read counts fromparams.hto_matrix_fileredwith the assignment of the best-matched method pair after donor matching
Optional output: Extracting donor-specific variants¶
Only when 1) best_method1 for the optimal match (best_method1 and best_method2) is vireo and 2) identification of donor-specific or discriminatory variants is enabled, then in folder donor_match/donor_match_[best_method1]_[best_method2]:
donor_specific_variants.csv: a list of donor-specific variantsdonor_specific_variants_upset.png: An upset plot showing the number of donor-specific variantsdonor_genotype_subset_by_default_matched.vcf: Donor genotypes of donor-specific variantsdonor_genotype_subset_by_vireo.vcf: Donor genotypes of a set of discriminatory variants filtered by Vireo